Predicting Drug-Target Interaction for New Drugs Using Enhanced Similarity Measures and Super-Target Clustering
Jian-Yu Shi a, Siu-Ming Yiu b, Yiming Li c, Henry C. M. Leung b, Francis Y. L. Chin *b
a School of Life Sciences, Northwestern Polytechnical University, No.127, Youyi Road West, Xi'an, Shaanxi, China, 710072
b Department of Computer Science, The University of Hong Kong, Pokfulam Road, Hong Kong
c Department of Psychiatry, The University of Hong Kong, Pokfulam Road, Hong Kong
INPUT
The INPUT to our method is the similarities between the new drug and those known drugs in the datasets .The drug similarity is measured by chemical-structure alignment and ATC, and the target similarity is measured by protein-sequence alignment and FC respectively. Both the similarities between the known drugs and those between the known targets are pre-calculated and built-in.
OUTPUT
The OUTPUT from our method is a set of the confidence scores which denote how likely this new drug interacts with all known targets respectively.
Computational Methods Involved
The computational methods used in our methods include Hattori et al.'s method [1] for drug chemical-structure similarity, the protein-sequence alignment algorithm of Smith and Waterman[2] for protein sequence similarity, the agglomerative hierarchical clustering [3] for building Super-Target and the KNN classifier [4] for performing DTI predictions.
The datasets used for training and evaluating our model were collected by Yamanishi et al [5] and can be downloaded from their webpage (http://web.kuicr.kyoto-u.ac.jp/supp/yoshi/drugtarget/). Yamanishi et al. [5] collected and integrated drug-target interactions from the KEGG BRITE [6], BRENDA [7], SuperTarget & Matador [8] and DrugBank databases [9]. These datasets were also used as the benchmark datasets for comparing the performance of DTI prediction algorithms by subsequent works (e.g. KBMF2K [10] and WNN-GIP [11]). The details about how to collect drugs, targets and their interactions, can be checked in their original paper [5].
All benchmark DTIs are split into four datasets according to the type of protein targets, including enzyme, ion channel (IC), G protein-coupled receptor (GPCR) and nuclear receptor (NR). The numbers of drugs, targets and their unknown interactions in the corresponding datasets are listed in Table 1.
| Enzyme | IC | GPCR | NR | |
| Number of drugs | 445 | 210 | 223 | 54 |
| Number of targets | 664 | 204 | 95 | 26 |
| Number of interactions | 2926 | 1476 | 635 | 90 |
All ATC codes of the drugs can be found in KEGG (http://www.kegg.jp/kegg/drug/) and all FC codes of the targets can be found in HUGO Gene Nomenclature Committee (http://www.genenames.org/).
Implementation
The input of our method includes pairwise drug similarities, pairwise target similarities and known drug-target interactions. The output is the confidence scores which denote how likely the pairs of known targets and new drugs are potential interactions. The whole workflow is illustrated in Fig 1.
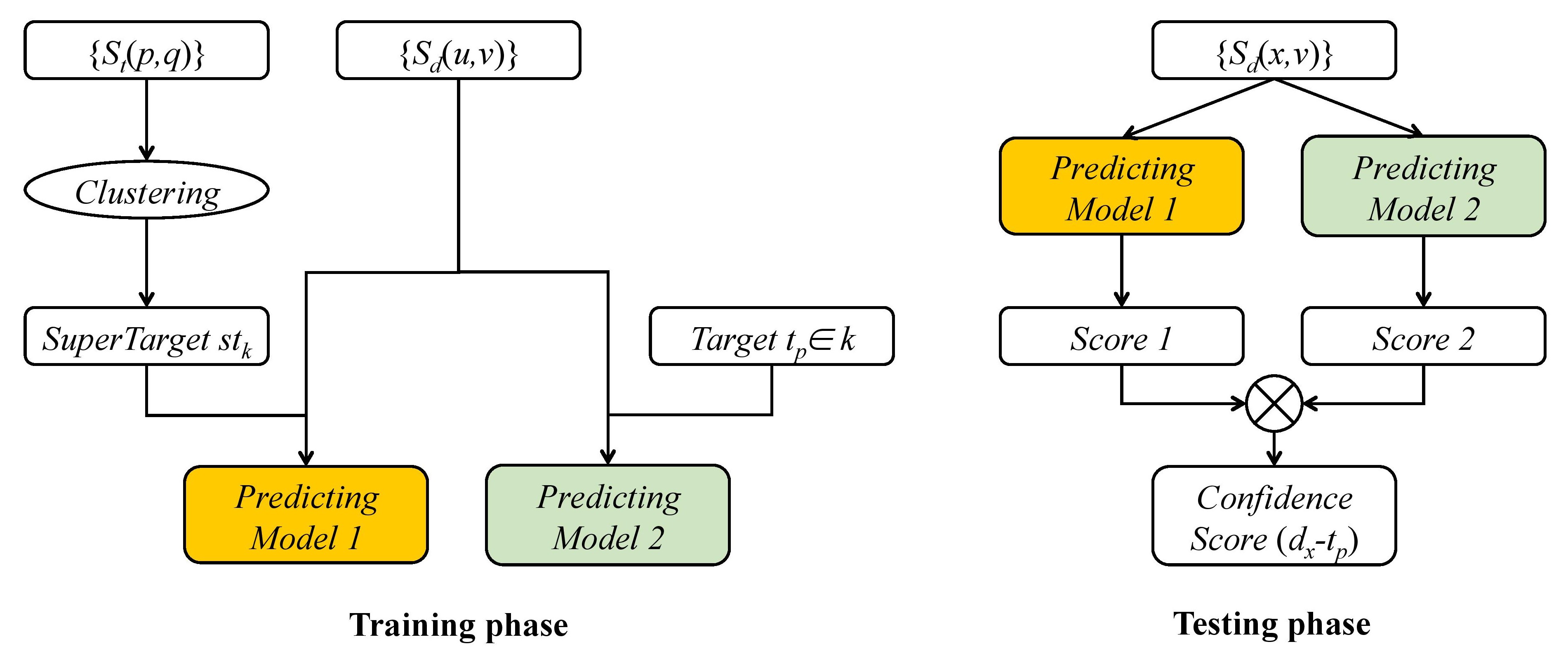 |
| Figure 1: The work ow of predicting drug-target interaction for new drugs. The whole work ow contains two phases,the training phase and the testing phase. In the first phase, pairwise similarities {Sd} between known drugs are used to build two predicting models for each super-target which is generated by clustering known targets based on pairwise target similarities {St}, and for each target in the super-target respectively. In the second phase, the similarities between a new drug dx and known drugs are input into Predicting Model 1 to obtain Score 1 which denotes how likely dx interacts with super-target stk, and then input into Predicting Model 2 to obtain Score 2 which re ects how likely dx interacts with the known target tp belonging to stk. The final confidence score of dx connecting with tp is obtained by the product of Score 1 and Score 2. Sd and St are the pairwise drug similarity and pairwise target similarity. |
Download: SourceCodes
References
- M. Hattori, Y. Okuno, S. Goto, et al., Development of a chemical structure comparison method for integrated analysis of chemical and genomic information in the metabolic pathways, J. Am. Chem. Soc. 125 (39) (2003) 11853-11865.
- T. F. Smith, M. S. Waterman, Identification of common molecular subsequences, J. Mol. Biol. 147 (1) (1981) 195-197.
- J. H. Ward Jr, Hierarchical grouping to optimize an objective function, J. Am. Statist. Assoc. 58 (301) (1963) 236-244.
- M.-L. Zhang, Z.-H. Zhou, ML-KNN: A lazy learning approach to multi-label learning, Pattern Recogn. 40 (7) (2007) 2038-2048.
- Y. Yamanishi, M. Araki, A. Gutteridge, et al., Prediction of drug-target interaction networks from the integration of chemical and genomic spaces, Bioinformatics 24 (13) (2008) i232-i240.
- M. Kanehisa, S. Goto, M. Hattori, et al., From genomics to chemical genomics: new developments in KEGG, Nucleic Acids Res. 34 (Database issue) (2006) D354-D357.
- I. Schomburg, A. Chang, C. Ebeling, et al., BRENDA, the enzyme database: updates and major new developments, Nucleic Acids Res. 32 (Database issue) (2004) D431-D433.
- S. Günther, M. Kuhn, M. Dunkel, et al., SuperTarget and Matador: resources for exploring drug-target relationships, Nucleic Acids Res. 36 (Database issue) (2008) D919-D922.
- D. S. Wishart, C. Knox, A. C. Guo, et al., DrugBank: a knowledgebase for drugs, drug actions and drug targets, Nucleic Acids Res. 36 (Database issue) (2008) D901-D906.
- M. Gönen, Predicting drug-target interactions from chemical and genomic kernels using Bayesian matrix factorization, Bioinformatics 28 (18) (2012) 2304-2310.
- T. van Laarhoven, E. Marchiori, Predicting drug-target interactions for new drug compounds using a weighted nearest neighbor profile, PloS One 8 (6) (2013) e66952.